The complete sequence of a human genome
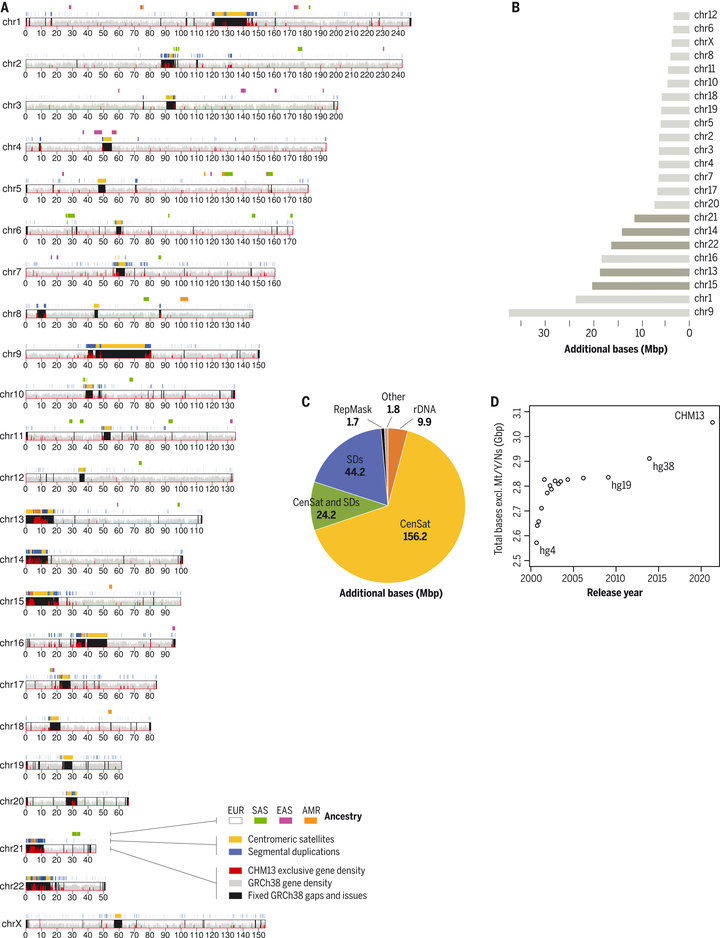 Image credit:
Science
Image credit:
Science
{kind=link}
Sergey Nurk et al. ,The complete sequence of a human genome. Science376,44-53(2022). DOI:10.1126/science.abj6987
Since its initial release in 2000, the human reference genome has covered only the euchromatic fraction of the genome, leaving important heterochromatic regions unfinished. Addressing the remaining 8% of the genome, the Telomere-to-Telomere (T2T) Consortium presents a complete 3.055 billion–base pair sequence of a human genome, T2T-CHM13, that includes gapless assemblies for all chromosomes except Y, corrects errors in the prior references, and introduces nearly 200 million base pairs of sequence containing 1956 gene predictions, 99 of which are predicted to be protein coding. The completed regions include all centromeric satellite arrays, recent segmental duplications, and the short arms of all five acrocentric chromosomes, unlocking these complex regions of the genome to variational and functional studies.
El artículo titulado “The complete sequence of a human genome” publicado en Science en 2022, presenta el trabajo del Consorcio Telomere-to-Telomere (T2T) que logró secuenciar completamente el genoma humano, incluyendo regiones previamente inaccesibles. La nueva referencia, T2T-CHM13, abarca 3.055 millones de pares de bases y corrige errores en referencias anteriores, añadiendo casi 200 millones de pares de bases con 1956 predicciones genéticas, de las cuales 99 son codificantes de proteínas. Este avance permite estudios más precisos sobre variaciones y funciones genómicas, abarcando todas las regiones centroméricas y los brazos cortos de los cinco cromosomas acrocéntricos.
Contexto y Limitaciones del Genoma de Referencia Anterior
El genoma de referencia humano previo, conocido como GRCh38, fue lanzado por el Consorcio de Referencia del Genoma (GRC) en 2013 y actualizado por última vez en 2019. Este genoma se originó a partir del Proyecto Genoma Humano y fue mejorado continuamente durante dos décadas. Sin embargo, presentaba limitaciones significativas debido a la subrepresentación de secuencias repetitivas y la presencia de haplotipos mosaicos, lo cual resultó en brechas no resolubles y regiones polimórficas incorrectamente ensambladas.
Avances Tecnológicos y Metodológicos
Para superar estas limitaciones, el Consorcio T2T utilizó tecnologías de secuenciación de lectura larga como PacBio HiFi y Oxford Nanopore ultralong-read. Estas tecnologías permitieron resolver variaciones estructurales complejas y brechas en GRCh38. La alta precisión de las lecturas HiFi, con una tasa de error del 0.1%, y la capacidad de las lecturas ultralong-read para abarcar repeticiones largas fueron fundamentales para el ensamblaje completo del genoma.
Resultados del Ensamblaje T2T-CHM13
El ensamblaje T2T-CHM13 elimina una barrera de 20 años que había ocultado el 8% del genoma, incluyendo todas las regiones centroméricas y los brazos cortos de cinco cromosomas humanos. El ensamblaje resultante es una secuencia completa y continua de un genoma humano que incluye todas las secuencias necesarias para procesos celulares fundamentales. Además, el nuevo ensamblaje corrige errores en referencias anteriores y añade secuencias previamente no alineadas con GRCh38.
Impacto y Aplicaciones Futuras
El genoma T2T-CHM13 proporciona una referencia más completa, precisa y representativa para estudios de variación genómica y funcionalidad en muestras humanas de todas las ascendencias. Este avance abre nuevas oportunidades para el estudio de la variación estructural y su asociación con fenotipos en regiones del genoma previamente inaccesibles. Además, el Consorcio T2T ha colaborado con el Consorcio de Referencia del Pan-Genoma Humano para construir una colección de haplotipos de referencia de alta calidad a partir de una muestra diversa de individuos, lo que permitirá un entendimiento más amplio de la diversidad genética humana.
Conclusión
El ensamblaje T2T-CHM13 representa un hito significativo en la genómica humana, proporcionando una referencia completa y precisa que superará las limitaciones de las referencias anteriores. Este avance permitirá estudios más detallados y precisos de la variación y funcionalidad genómica, impulsando futuros descubrimientos en la salud y enfermedades humanas.
Referencias: