Trabajo Práctico 3 - Análisis transcriptómico por RNA-Seq

Objetivos:
El objetivo de este trabajo práctico es introducir a los alumnos en el análisis de datos transcriptómicos generados en experimentos de RNA-Seq partiendo de archivos fastq hasta llegar a la matriz de expresión. Debido al tiempo disponible para la realización de esta práctica no se hará énfasis en el entendimiento de los códigos de programación, si no que se buscará que comprendan los conceptos más importantes que hay detrás de cada paso del análisis bioinformático.
Introducción:
La transcriptómica implica el estudio a gran escala de las moléculas de ARN expresadas por las células en un momento determinado. El avance de la tecnología permitió pasar de estudiar la expresión de genes individuales al análisis de todo el ARN expresado por las células (transcriptoma). A diferencia de lo que pasa con el ADN que es, en su mayor parte, idéntico en todas las células, el ARN que se transcribe activamente en las células es dinámico, permitiendo un mayor entendimiento de los procesos biológicos que suceden en células y condiciones específicas, y de los mecanismos de regulación de estos. Esta técnica permite también dilucidar la estructura, modificaciones, y variaciones de transcriptos individuales en distintos contextos. De esta manera, la transcriptómica es actualmente una herramienta fundamental de la genómica funcional, un campo de estudio que intenta integrar grandes cantidades de datos con el objeto de entender los mecanismos que controlan el fenotipo celular, y en última instancia, el del organismo completo (Cieślik and Chinnaiyan, 2017).
Las herramientas para el estudio de la expresión de ARN han evolucionado con el paso del tiempo, desde la aparición de las técnicas de northern blot (Alwine, Kemp and Stark, 1977) hasta llegar a las tecnologías de nueva generación de las que disponemos hoy en día. El aumento en la cantidad de secuencias conocidas y la aparición de las técnicas para generar ADNc, han permitido el desarrollo de macro y microarreglos (microarrays). Este fue un paso fundamental en el campo de la transcriptómica que permitió aumentar la cantidad de genes a analizar y disminuir los costos. Hasta el día de hoy los microarrays siguen siendo utilizados debido al amplio conocimiento y la experiencia que se tiene en su uso, aunque desde la aparición de las tecnologías de secuenciación de nueva generación (NGS) han empezado a dejarle lugar a las nuevas tecnologías que han reducido los costos y aumentado la eficiencia de manera dramática. Es por ese motivo que en este trabajo práctico nos centraremos en el análisis de datos de RNA-Seq.
Secuenciación transcriptómica RNA-Seq
El RNA-Seq es una técnica de nueva generación que utiliza las nuevas tecnologías de secuenciación de ADN para el análisis transcriptómico. Previo a la secuenciación, se debe aislar y purificar el ARN de la muestra a analizar. Existe una gran variedad de protocolos para realizarlo dependiendo de la fuente desde la cual se hará la extracción y de las preguntas biológicas a analizar (Mortazavi et al., 2008). Por ejemplo, en el caso de querer analizar solo el ARNm se realizan purificaciones específicas para obtener solo estas moléculas de ARN. Los protocolos también toman cuenta de la fragilidad del ARN en comparación con el ADN, además de su susceptibilidad a ser degradado por ribonucleasas. Luego de la purificación, es necesario fragmentar el ARN para obtener las longitudes óptimas según el secuenciador a utilizar, hacer la síntesis de ADNc y adicionar los adaptadores de secuenciación tal como se hace para la secuenciación de ADN genómico. Luego de secuenciar obtendremos los archivos fastq, que son archivos de texto plano que contienen las secuencias correspondientes a cada una de las lecturas obtenidas. En el caso de experimentos de secuenciación single-end tendremos un archivo fastq por muestra. En cambio, si se realizó la secuenciación paired-end tendremos dos archivos por muestra, uno con las lecturas forward y otro con las reverse.
Análisis bioinformático en RNA-Seq - Primera Parte
Una vez obtenidos los archivos fastq se debe realizar el análisis bioinformático para obtener información que nos permita interpretar biológicamente los resultados:
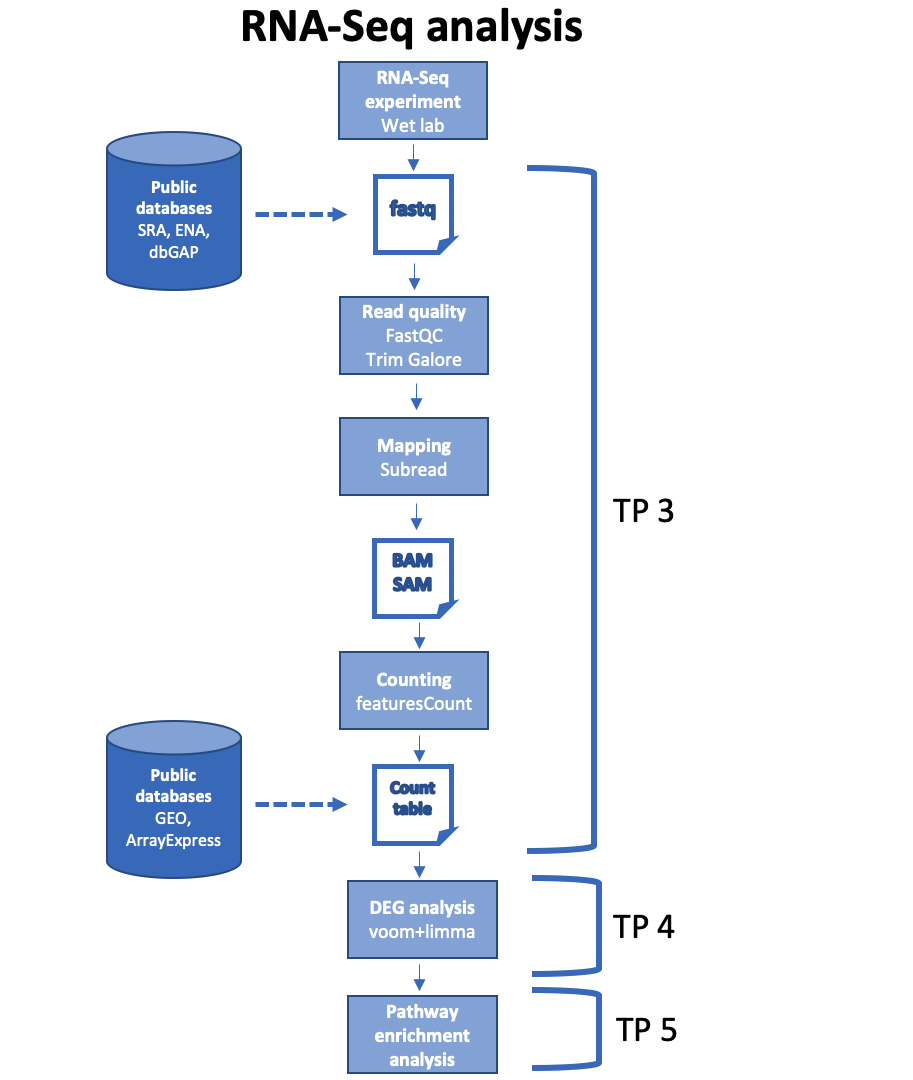 Figura 1
Figura 1
En primer lugar se debe proceder con el control de calidad de las lecturas. Los archivos fastq además de contener la secuencia nucleótidos de cada lectura, contienen información de la calidad con la que el secuenciador leyó cada posición. De esta manera, a partir de programas bioinformáticos como el FastQC, es posible analizar y hacer un control de la calidad de los datos. Luego, en base a este análisis, se realiza el llamado trimming de los fastq, que consiste principalmente en la eliminación de las lecturas de mala calidad, recorte de las lecturas (especialmente hacia el final de las lecturas la calidad suele decaer y es conveniente recortarlas), y eliminación de secuencias asociadas a los adaptadores utilizados durante la secuenciación.
Una vez que se realizó el preprocesamiento de los archivos fastq se procede a alinear las lecturas contra el genoma de referencia o contra un transcriptoma. El procedimiento puede variar en base al pipeline utilizado. Lo más común en el caso de RNA-Seq humano o de ratón es alinear contra el genoma de referencia utilizando programas de alineamiento especialmente desarrollados para la secuenciación de ARNm, ya que debe tener en cuenta los sitios de splicing e intrones a la hora de alinear. En ese caso se obtiene como resultado un archivo BAM. Este archivo contiene toda la información de las lecturas y del alineamiento contra el genoma de referencia. Luego, se utilizan programas para contabilizar la cantidad de lecturas que mapearon contra cada gen, de manera de obtener una tabla de counts o matriz de expresión.
Por otro lado, en los últimos años han surgidos nuevos métodos para cuantificar la expresión de transcriptos a partir de datos de RNA-Seq que hacen lo que se denomina “pseudomapeo” o “pseudoalineamiento”. Este tipo de métodos evitan el alineamiento de lecturas con el genoma de referencia y, en su lugar, se mapean la lecturas a un conjunto de transcritos compatibles buscando coincidencias entre kmers de las lecturas y de un índice de transcriptos. Estos métodos han demostrado ser mucho más rápidos y eficientes computacionalmente (se pueden correr en computadoras de escritorio comunes) sin perder precisión. Ejemplos de este tipo de programas son Kallisto y Salmon. En este TP usaremos Kallisto para cuantificar los niveles de expresión de nuestros datos de RNA-Seq.
La estructura de la tabla que obtenemos finalmente con los distintos métodos es la misma, genes en filas y muestras en columnas:
| Gene | Sample1 | Sample2 | SampleN |
|---|---|---|---|
| Gene1 | 4 | 5 | … |
| Gene2 | 3 | 0 | … |
| GeneN | … | … | … |
Esta tabla sirve de input para infinidad de pipelines y softwares que nos van a permitir obtener e interpretar la información biológica obtenida del experimento. Algunos ejemplos:
- Análisis de expresión diferencial: comparar la expresión génica de un grupo de muestras contra otro, por ejemplo tumor vs tejido normal, o un estímulo vs control.
- Clustering: Esto permite agrupar las muestras de manera no supervisada y encontrar subgrupos con un patrón de expresión génica similar.
- Expresión de marcadores: Se puede análizar la expresión de genes particulares o de firmas genéticas más grandes y buscar asociaciones con las variables de mi experimento. Por ejemplo, se puede estudiar una firma genética para ver si correlaciona con la respuesta a un tratamiento, con la sobrevida de los pacientes o con algún estímulo que se esté estudiando.
- Análisis funcionales (enriquecimiento de pathways o vías biológicas, o de set de genes con funciones similares): A partir de los resultados del análisis de expresión diferencial se puede estudiar que vías o pathways están desregulados en la condición de estudio. Por ejemplo, si estamos comparando células tratadas con una droga vs un control, podemos ver los pathways que están siendo alterados por el tratamiento.
- Análisis de redes de expresión génica: A partir de los datos transcriptómicos se pueden inferir módulos de genes que se coexpresan. El estudio de los mecanismos regulatorios comunes a los genes de cada modulo permite inferir la actividad de factores de transcripción que estén activando o reprimiendolos bajo distintas condiciones.
- Análisis de deconvolución: Este tipo de análisis permiten estimar las poblaciones celulares presentes en una muestra a partir de la transcriptómica total. Por ejemplo, si análizamos la transcriptómica de una biopsia tumoral es posible inferir la proporción de distintos tipos celulares del sistema inmune que están infiltrando el tumor (es decir, que estan presentes dentro del tumor). De esta manera es posible estudiar la respuesta inmune en distintos tipos de tumor y bajo distintas condiciones de tratamiento y respuesta a drogas, entre otras cosas.
Bases de datos públicas
Debido a la disminución en los costos de secuenciación la tecnología de RNA-Seq es cada vez más accesible. Esto, sumado a la gran cantidad de información que brinda, hace que este tipo de estudios sean ampliamante utilizados en todos los laboratorios del mundo. La gran mayoría de los papers que publican resultados con datos de RNA-Seq dejan depositados los datos en bases públicas para que puedan ser reutilizados por la comunidad científica (muchas revistas lo exigen). Esto nos brinda una fuente de recursos extraordinario para obtener información que nos permita generar nuevas hipótesis o validar nuestros hallazgos en estudios in vivo o in vitro en el laboratorio. Por ejemplo, de esta manera podemos analizar datos de grandes cohortes de pacientes, cosa que sería muy díficil y costoso de realizar de otra manera.
Estos datos pueden estar en distinto formato y grado de preprocesamiento. Lo ideal, y lo que ocurre en la mayoría de los casos, es que los datos del experimento estén dsiponibles en formato de tabla de counts. Esto nos permite bajar la tabla de expresión y utilizarla como input para infinidad de análisis. Además estos datos ocupan poco espacio y en general es posible analizarlos desde computadoras personales de mediana capacidad. Este tipo de datos los autores de los trabajos suelen subirlos a Gene Expression Omnibus (GEO) que es parte del NCBI. También es posible encontrar algunos trabajos depositados solamente en ArrayExpress, que pertenece al EMBL-EBI europeo. En muchos casos se encuentrar subidos en ambos repositorios. Es posible que en algunos casos en vez de la tabla de counts tengamos la tabla normalizada en TPM (Transcripts per Million) o FPKM (Fragments per Kilobase per Million). Estos formatos normalizan las counts por el tamaños de la librería (cantidad de lecturas secuenciadas en cada muestra) y por el largo de cada gen (cuanto más largo el gen más lecturas se espera que alineen contra él). Estos formatos son útiles para visualizar los datos en heatmaps o clustering, pero no son los indicados para hacer análisis de expresión diferencial.
En algunos pocos casos es posible que no estén los datos preprocesados y solo se tenga acceso a los archivos fastq. En este caso la única alternativa es realizar todo el análisis desde cero. El problema en ese casos es la capacidad computacional necesaria para realizarlo. Los archivos crudos son extremadamente grandes y se necesitan máquinas con múltiples procesadores y una gran cantidad de memoria RAM para procesarlos. Esto se suele realizar en clusters de computadoras o servidores especializados para este tipo de análisis. Además, en algunos casos los datos de secuenciación crudos en fastq pueden estar con acceso controlado. Es decir que como tienen los datos de las secuencias, estos pueden estar protegidos por acuerdos de confidencialidad y es necesario pedir un permiso para descargarlos y utilizarlos. Esto no es necesario cuando los datos se encuentran publicados en un formato ya preprocesado como tablas de expresión.
Importante: Todo el pipeline de análisis de RNA-Seq se encuentra dividido en tres trabajos prácticos. En el TP3 veremos el análisis desde los fastq hasta obtener la tabla de expresión en counts. En el TP4 realizaremos el análisis de expresión diferencial entre las dos condiciones del estudio, lo que permitirá ver los genes que se encuentran up o down-regulados en cada una. Por último, en el TP5 realizaremos análisis de enriquecimiento pathways y de set de genes para poder interpretar biológicamente los resultados obtenidos. Se deberá realizar un único informe para estos tres trabajos prácticos. A lo largo de los TPs verán preguntas que les servirán como guía para la realización del informe. Las instrucciones detalladas para realizarlo se les dará durante la clase.
Resumen del trabajo práctico: Análisis transcriptómico de pacientes con COVID19
En esta primer parte del trabajo práctico de transcriptómica realizaremos todo el pipeline de análisis que va desde los archivos fastq hasta obtener la matriz de expresión o tabla de counts. Para ello, utilizaremos la VM que posee los programas necesarios instalados. Los archivos que utilizaremos se encuentran en la carpeta TP3.
Para este trabajo analizaremos muestras de pulmón de pacientes infectados con SARS-CoV-2 y donantes no infectados como control. Las muestras corresponden al estudio publicado en la revista Cell en el mes de mayo del 2020: “Imbalanced Host Response to SARS-CoV2 Drives Development of COVID19 (Blanco-Melo et al., 2020) Link: https://doi.org/10.1016/j.cell.2020.04.026:
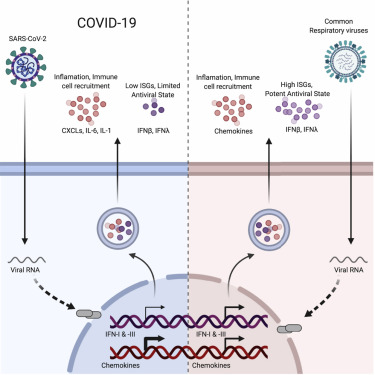
Graphical abstract de Blanco-Melo et al., 2020.
Por una cuestión de tiempo y de capacidad computacional se utilizará un set de datos reducido que solo contiene las lecturas correspondientes al cromosoma 21 de cada muestra. En un análisis real el pipeline sería exactamente igual, solo que se utilizarían los archivos fastq completos y se mapearía contra el transcriptoma de referencia entero. En este caso, como solo tenemos las lecturas filtradas solo lo haremos contra los transcriptos del cromosoma 21. La idea es estudiar los genes que están up o down- regulados durante la infección del virus SARS-CoV-2 y encontrar los pathways o vías biológicas desreguladas durante la infección. Esta información nos permite describir mejor los procesos biológicos subyacentes y plantear nuevas hipótesis de trabajo. También resulta de vital importancia a la hora de buscar nuevos tratamientos para los pacientes.
Desarrollo:
Para este TP se debe utilizar la máquina virtual Ubuntu provista por los docentes.
Nota: Recordar activar el environment genomica_uade de conda antes de empezar.
En primer lugar vamos a abrir la terminal y nos moveremos a la carpeta del TP3.
cd ./TPs/TP3
ls -l
- Qué archivos hay en la carpeta?
Vamos a analizar en primer lugar la calidad de las lecturas en nuestros fastq con el programa FastQC. Primero, creamos una carpeta para guardar los resultados del informe de calidad y luego corremos el programa sobre todas las muestras.
mkdir FASTQC
fastqc --threads 2 -outdir ./FASTQC Series*
Los archivos del informe del FastQC se encuentran en la carpeta FASTQC en formato html. Pueden navegar hacia la carpeta desde la interfaz visual de Ubuntu y al hacer doble click en el archivo se les abrirá en el navegador de internet.
- Cuál es su opinión acerca de la calidad de las lecturas? (Tengan en cuenta que estos archivos fastq ya fueron trimmeados cuando se hizo el subset de lecturas del cromosoma 21 para este TP).
El siguiente paso es hacer el trimming de las lecturas con el programa Trim Galore. Utilizaremos los parámetros por defecto, que utilizan un threshold para la calidad de las lecturas de 20. También se recortan las bases de las lecturas en los extremos si tienen baja calidad. Primero crearemos una carpeta en donde guardar las salidas de los programas que usaremos y luego utilizamos nuevamente un loop for para procesar las 4 muestras.
mkdir Results
for i in Series*; do
trim_galore --cores 2 $i \
-o ./Results/
done
En la carpeta Results van a encontrar los archivos fastq nuevos ya trimmeados que se usarán para el alineamiento. Además encontraran archivos de reporte con el informe del procesamiento con Trim Galore.
- Qué información tienen estos reportes?
Pueden leerlos de la siguiente manera:
cat Results/Series15_HealthyLungBiopsy_1_chr21.fastq_trimming_report.txt
Puede repetirlo con las demás muestras.
Ahora pasaremos a mapear las lecturas contra el transcriptoma de referencia. En este caso, como solo trabajamos con las lecturas correspondientes al cromosoma 21, haremos el mapeo solamente contra ese cromosoma. El archvio fasta con las secuencias de los transcriptos (cDNA) del cromosoma 21 y el archivo .gtf con la anoración del mismo (es decir, la información sobre los genes que están en cada posisicón genómica) se encuentran en la carpeta Refs. Todo esto se hará con Kallisto que, como comentamos anteriormente, realiza un pseudomapeo de cada lectura buscando los k-mers en la misma y luego buscándolos en la tabla de índices hash para identificar los transcriptos compatibles (Ver manual en la bibliografía al final de la guía). Por lo tanto, el resultado final del programa es el conteo de lecturas mapeadas contra cada transcripto. Si bien a veces se buscar realizar análisis de expresión diferencial de transcriptos entre distintas condiciones, la mayoría de las veces se busca ver la expresión diferencial a nivel de genes. Por lo tanto, en la parte final del TP vamos a importar los resultados de Kallisto a R y vamos a generar una tabla final de counts por gen sumando los valores de cada transcripto correspondiente al mismo gen. Para ello usaremos RStudio, un environment de desarrollo para trabajar con el lenguaje R. Si bien lo empezaremos a usar al final de este TP, lo aprenderemos a usar con un poco más de profundidad en el próximo trabajo práctico.
Nota: Tener en cuenta que hay distintas versiones del genoma/transcriptoma humano. Siempre utilizar la misma versión para todos los análisis (se recomienda usar la última versión, aunque en ciertos casos pueden necesitar utilizar una versióna anterior). Además, para cada actualización del genoma existen distintas versiones del ensamblado que difieren ligeramente. En este caso también se debe utilizar siempre el mismo ensamblado para todo el análisis ya que puede haber discrepancias en el formato. Principalmente existen dos versiones, una que es la del Consorcio Internacional del Genoma Humano (última versión GRCh38) y la de la Universidad de California Santa Cruz (UCSC, última versión nombrada hg38). En este caso utilizamos el transcriptoma de la versión GRCh38, la última versión descargada de la base de datos de Ensembl. Es importante destacar, que distintas versiones del genoma de referencia descargadas de distintas bases de datos cuentan con diferencias en cómo se nombran los cromosomas (chr1 vs 1), por lo cuál mezclar anotaciones de distintas fuentes puede generar problemas durante el análisis de los datos.
En primer lugar debemos generar un índice del genoma (chr21):
kallisto index -i ./Refs/HS.GRCh38.cdna.21.idx ./Refs/Homo_sapiens.GRCh38.cdna.21.fa.gz
Luego de correr el programa se les generará un archivo en la carpeta Refs que corresponde al transcriptoma indexado. Este archivo lo utilizará el programa que realiza el mapeo.
Para el pseudomapping deberemos pasarle al programa el índice del transcriptoma que generamos en el paso anterior y los archivos fastq trimmeados. Creamos primero una carpeta para guardar los resultados:
mkdir kallisto_output
El archivo sample_names.txt tiene el nombre de cada muestra una abajo de la otra. Este código es para correr los FASTQ trimmeados con Trim-Galore:
while read i; do
kallisto quant -i ./Refs/HS.GRCh38.cdna.21.idx -t 2 --single -l 200 -s 20 Results/${i}_trimmed.fq -o ./kallisto_output/${i}
done < ./sample_names.txt
-
Mirando la salida del programa en la terminal. ¿Qué porcentaje de las lecturas se mapearon correctamente en cada muestra?
-
Observe los archivos generados. ¿Qué tiene cada archivo? ¿Qué tiene cada columna de los archivos abundance.tsv?
Una vez que tenemos los archivos generados por Kallisto podemos pasar a R, en donde haremos los análisis de los datos en los próximos TPs. Vamos a finalizar esta parte del pipeline importando los datos a un proyecto de R con el paquete tximport. Este paquete importa los archivos generados por Kallisto, los unifica en una sola tabla para todas las muestras, y nos permite sumar las counts de los transcriptos correspondientes al mismo gen para tener una tabla final con counts de genes para cada muestra.
Nota: Como mencionamos anteriormente, si bien empezaremos a usar R y RStudio en esta última parte del TP, aprenderemos un poco más de ambos en el próximo trabajo práctico.
-
Primero abrir RStudio y crear un nuevo proyecto en el carpeta TP3.
-
Abrir un nuevo R script y luego pegar el siguiente código. Para ejecutarlo se pueden parar sobre cada comando y hacer ctrl+enter en cada uno.
#Cargamos las librerías necesarias
library(ensembldb)
library(AnnotationHub)
library(tximport)
library(dplyr)
#Cargamos la base de datos de Ensembl para obtener la info de los transcriptos de cada gen
ah <- AnnotationHub()
ahDb <- query(ah, "EnsDb.Hsapiens.v107")
ahEdb <- ahDb[[1]]
#Generamos la tabla de anotacion de transcripto a gen
k <- keys(ahEdb, keytype = "TXNAME")
tx2gene_annotation <- select(ahEdb, k,c("TXIDVERSION","GENEIDVERSION","GENENAME"),"TXNAME")
tx2gene <- tx2gene_annotation[,c(2,3)]
#Generamos un listado de los path a cada archivo abundance.h5 de cada muestra
samples <- c("Series15_COVID19Lung_1_chr21",
"Series15_COVID19Lung_2_chr21",
"Series15_HealthyLungBiopsy_1_chr21",
"Series15_HealthyLungBiopsy_2_chr21")
dir <- "./kallisto_output"
files <- file.path(dir, samples,"abundance.h5")
names(files) <- samples
#Importamos los datos a R con tximport
txi <- tximport(files, type = "kallisto",tx2gene = tx2gene, countsFromAbundance = "lengthScaledTPM")
names(txi)
#Extraemos los TPM y les cambiamos los Ensembl IDs por los Gene Symbol
txi_tpm <- txi$abundance
txi_tpm_rownames <- data.frame(GENEIDVERSION = rownames(txi_tpm))
txi_tpm_rownames <- merge(txi_tpm_rownames, tx2gene_annotation[,c(3,4)], all.x=T)
txi_tpm_rownames <- distinct(txi_tpm_rownames)
rownames(c) <- txi_tpm_rownames$GENENAME
#Extraemos las counts
txi_counts <- txi$counts
#Visualizar tabla final de TPMs
View(txi_import)
#Visualizar tabla final de counts
View(txi_counts)
- Mirando las tablas finales, ¿Qué diferencias hay entre una y otra? ¿Qué estructura tienes las tablas? ¿Qué muestra cada columna?
Llegado este punto ya tenemos la contabilización de las lecturas que mapearon contra cada gen del transcriptoma de referencia (chr 21 en este caso).
Nota: La tabla final de counts obtenida muestra tiene los IDs de la base de datos Ensembl para cada gen. Muchos de los programas bioinformáticos que utilizan tablas de counts como input pueden utilizar tanto los IDs de Ensembl como los gene symbols para identificar a los genes. Sin embargo, algunos programos solo trabajan con un tipo particular de identifiador. Algunos programas utilizan los IDs de Entrez para identificar a los genes, en ese caso habra que utilizar alguna herramienta que nos traduzca alguno de estos tipos de IDs a los de Entrez (para esto se puede usar biomRt desde la web de Ensembl o su paquete para R).
Felicidades!! Con esto terminamos la primera parte del pipeline de análisis de datos de RNA-Seq. Comenzando desde los datos crudos fastq llegamos a obtener la tabla de expresión en counts que nos servirá para los análisis subsiguientes.
Bibliografía
- Cieślik, M., Chinnaiyan, A. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet 19, 93–109 (2018). https://doi.org/10.1038/nrg.2017.96
- J C Alwine, D J Kemp, G R Stark. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. PNAS Dec 1977, 74 (12) 5350-5354; DOI: 10.1073/pnas.74.12.5350
- Mortazavi, A., Williams, B., McCue, K. et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628 (2008). https://doi.org/10.1038/nmeth.1226
- https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- https://pachterlab.github.io/kallisto/
- Blanco-Melo et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020 181-5 1036–1045.e9 doi: 10.1016/j.cell.2020.04.026