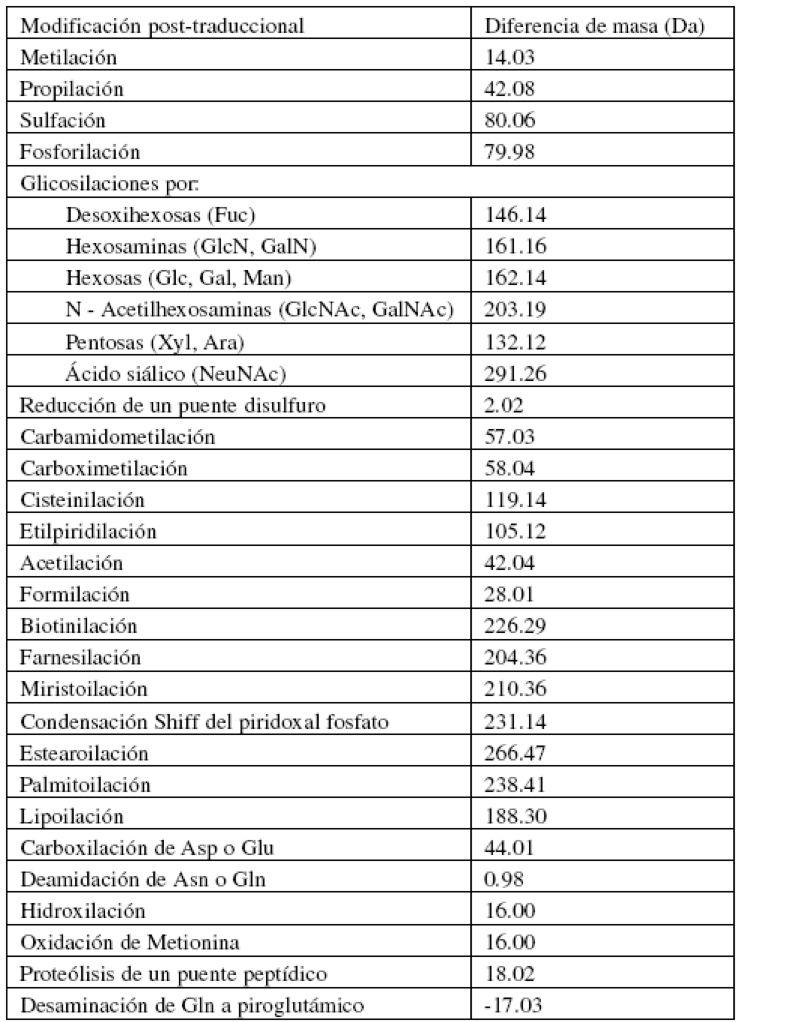
Trabajo Práctico 6 - Identificación de proteínas por espectometría de masas

Utilización de software para análisis de datos de espectrometría de masa de proteínas: Identificación de proteínas por el método de la huella peptídica (peptide mass fingerprinting o PMF) y con fragmentación MS/MS.
Objetivos:
- Llevar a cabo una identificación de proteína por el método de la huella peptídica, utilizando un programa disponible en Internet: Mascot.
- Utilizar datos de fragmentación de proteínas por MS/MS para complementar y aumentar la confiabilidad de la identificación.
- Evaluar el efecto de distintas variables experimentales a tener en cuenta durante la identificación.
Link
MASCOT: http://www.matrixscience.com/search_form_select.html
Introducción
En este TP veremos cómo a partir de datos de espectrometría de masas nosotros podemos determinar, con una cierta confiabilidad,la identidad de una proteína. Para ello haremos uso de las técnicas denominada Peptide Mass Fingerprinting (PMF, en castellano “huella peptidica”) y Peptide Fragment Fingerprinting (PFF). La técnica de huella peptídica se basa en determinar la masa molecular exacta de péptidos derivados de una proteína purificada. Dichos péptidos son obtenidos a partir de una digestión enzimática con tripsina, que corta a la proteína en sitios bien definidos: luego de una lisina (K) o una arginina (R). La lista de masas moleculares de los péptidos digeridos se compara con una digestión «virtual» que un motor de búsqueda hace sobre una base de datos pública, utilizando el criterio de corte de la enzima utilizada. La proteína se considera identificada cuando tiene «picos» en común con alguna proteína de la base de datos y un score significativo.
¿Qué datos hay que proveerle al motor de búsqueda?
- Lista de masas moleculares obtenidas experimentalmente a partir del análisis MS de un digerido de la proteína. La misma puede incluir las intensidades de los picos.
- Descripción del procesamiento de la muestra
- Modificaciones químicas introducidas (generalmente reducción/alquilación)
- Nombre de la enzima utilizada para digerir la muestra (generalmente tripsina)
- Características de exactitud del instrumento utilizado (lo que se denomina « mass o peptide tolerance »)
- Características del espacio de búsqueda
- Base de datos a utilizar
- Restricciones taxonómicas
- Restricciones de pI y/o MW
¿Qué nos devuelve como resultado el motor de búsqueda?
- Un número de acceso para una o más proteínas que representan el resultado de la identificación
- Un score que da idea de cuán confiable es la determinación de identidad, asi como otros parámetros que permiten controlar la calidad de la identificación (distribución de errores, etc.)
- Una lista detallada de potenciales péptidos asignados
- Una lista de péptidos no asignados
- Un resumen de los parámetros de búsqueda utilizados
MASCOT
Mascot es un motor de búsqueda desarrollado por un grupo de investigadores del Imperial Cancer Research Fund de Londres, que fue comercializado por la empresa Matrix Science. La misma provee al público de una versión online con capacidades limitadas en cuanto a procesamiento de grandes volúmenes de datos, pero suficientes para las consultas habituales. Mascot permite procesar datos espectrométricos para huella peptídica (PMF), identificación PFF (con datos de fragmentación), y secuenciación de novo, que son las 3 formas de secuenciación de péptidos posibles de realizar con datos de espectrometría de masa. Nosotros vamos a trabajar con datos provenientes de la digestión tríptica de un spot de un gel bidimensional, que fueron obtenidos con un equipo MALDI-TOF/TOF. Previo a la digestión, la muestra fue tratada con un agente reductor (DTT, ditiotreitol) y un agente alquilante, la iodoacetamida. Este tratamiento incorpora un residuo carbamidometilo a las cisteínas (Cys) presentes en la proteína a analizar, previniendo la formacion de puentes disulfuro intra o intermoleculares que impedirían la secuenciación de péptidos con Cys.
**Busque en Google la palabra Mascot. Vaya al sitio de Matrix Science. **
En esa pantalla pique el hipertexto Perform Search en la opción Peptide Mass Fingerprint. Le aparecerá una pantalla con opciones. Elija la que dice Peptide Mass Fingerprinting. La pantalla para huella peptídica del Mascot es la siguiente:
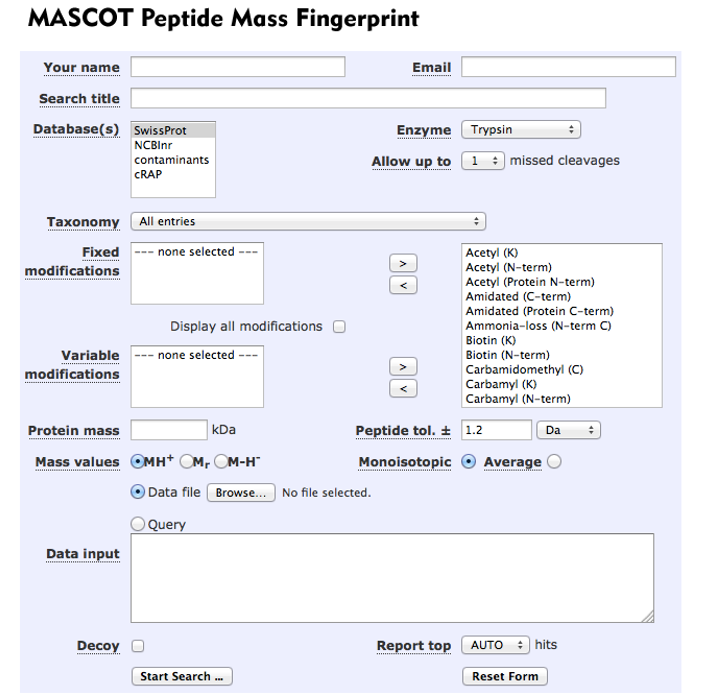
Se observan las siguientes categorías:
-
**Name, E-mail**: siempre se debe completar este campo con un nombre y una direccion de mail válida porque si no no es aceptada la búsqueda. -
Search Title: opcional
-
Database: es la base de datos de secuencias que se utilizará para la digestión virtual con la proteasa de elección. Podemos usar la base de datos curada de SwissProt o la NCBInr (non-restricted), que incluye a todos los datos depositados en el NCBI.
-
Taxonomy: se selecciona si queremos buscar sólo proteínas humanas, o de mamíferos o de todos los taxones.
-
Enzyme: se elige la enzima que se utilizó para digerir la muestra experimental. Generalmente es la tripsina
-
Allow up to: Esta opción permite considerar que existen péptidos derivados de digestiones incompletas de la enzima proteolítica, que también pueden ser evaluados. Generalmente se considera probable hasta un clivaje perdido.
-
Fixed and Variable modifications: son las modificaciones postraduccionales introducidas experimentalmente (fixed), como la carbamidometilación de cisteínas, o las comunes en la naturaleza en al menos una subpoblación de las moléculas analizadas (variable). Se seleccionan mediante las flechas.
-
Protein mass (kDa). Si se desea se puede poner un techo al tamaño de la molécula de donde se obtuvieron los péptidos. Se usa poco.
-
Peptide tol. ±: error intrínseco de la medición, dependiente del espectrómetro. Esta tolerancia surge de las características del instrumento y de la calibración que se haya realizado. En general, cada muestra medida es calibrada internamente aprovechando 2 o 3 picos de autodigestión de la tripsina, que aparecen siempre con buena intensidad (casi siempre los de 842.5100 y 2211.1046)
-
Mass values: MH+ (default) es la comúnmente generada por MALDI (equivale al tamaño del péptido más un protón).
-
Monoisotopic or Average: se usa Monoisotopic si el instrumento utilizado permite determinar la composición isotópica.
-
Query: se selecciona esta opción cuando se le va a pastear una lista de masas. Alternativamente, se pueden insertar desde un archivo con la opción Data file.
-
Data input: aquí se inserta la lista de masas peptídicas experimentales, con o sin las intensidades asociadas.
-
Decoy: cuando esta opción está tickeada, la búsqueda se hace simultáneamente en la base de datos elegida Y en una base de datos DECOY formada por los mismos nucleótidos pero ordenados al azar. Permite distinguir mejor si los hits obtenidos son reales o pueden corresponder a asignaciones azarosas. Con los resultados se puede calcular el FDR (false discovery rate), definido como el:
Pept.Sig/Pept.Real x 100
Pept.Sig: número de péptidos significativos en la base decoy
Pept.Real: número de péptidos significativos en la base real
Este número no debería ser mayor al 1%
-
Report top: se refiere a cuántos “hits” quiero que informe. Los ordenará según score decreciente.
Una vez introducidos todos los parámetros, se clickea el Start Search. La pantalla de resultados default, denominada « Concise Protein Summary » luce similar a esta:

Se observa una primera parte que indica algunos de los parámetros seleccionados, y el nombre de la proteína que sale con el mejor score. Le sigue una tablita en la que vemos comparados los scores del primer hit, tanto en la base de datos original como en la base de datos decoy. Luego viene un gráfico que indica visualmente cuán significativo es el score. Presten atención a la posición sobre el eje X de la barra roja, y a su altura. Luego viene la lista de hits, separada por «familia de hits similares » Hacia el final de la lista de hits se encuentra un listado del resto de los parámetros seleccionados para la búsqueda. Hablaremos exhaustivamente de los resultados durante la clase.
El Score del Mascot
Mascot calcula un Score probabilístico que se basa en el algoritmo Mowse (Pappin et al, Curr Biol. 1993 Jun 1;3(6):327-32). Según este procedimiento, primero se observan cuáles de las masas de péptidos obtenidos a partir de la base de datos coinciden con las experimentales. Luego, para las masas que concuerdan (match), Mowse usa factores empíricos que ponderan cada uno de los matcheos. Por ejemplo, considera la abundancia relativa de péptidos de la misma masa en base de datos y compensa por el tamaño de la proteína. De esta manera calcula un número P que, para cada entrada de la base de datos que concuerde con los datos experimentales de la proteína desconocida, representa la probabilidad de que el matching observado sea un evento al azar y no una asignación válida. Ese número, P, es transformado en el Protein Score del Mascot mediante una transformación simple: -10(log)P. Entonces, una asignación que tiene una probabilidad de 10-8 de ser al azar se convierte en un Score de 80. Además, Mascot da un nivel de significación para este Score, que depende del tamaño de la base de datos chequeada (¿de qué depende el tamaño de la base de datos chequeada?). Para ello, calcula un umbral o threshold de identidad que representa el Score que tendría un evento que ocurriera al azar en el 5% de los casos. Si el valor de Score de la proteína supera el umbral, el Score es significativo con un p<0.05.
Para más info: http://www.matrixscience.com/help/scoring_help.html
IDENTIFICACION DE UNA PROTEÍNA DE UN SPOT DE GEL 2D POR HUELLA PEPTÍDICA
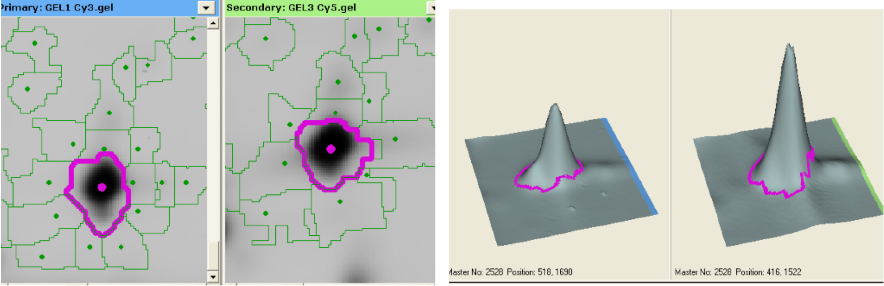
Este spot proviene de geles realizados con el secretoma de líneas celulares de melanoma humano con distinto grado de agresividad. En las dos líneas celulares, dicho spot está diferencial, siendo una imagen representativa la siguiente:
Se realizó una digestión in-gel con tripsina, previa reducción con DTT y alquilación de Cys con iodoacetamida (la cual genera la modificación carbamidometilación). Se eluyeron los péptidos y se cristalizaron junto con ácido sinapínico (matriz) en una placa de MALDI. Se analizaron en un MALDI-TOF/TOF en modo MS reflectrón, con calibración interna utilizando picos de tripsina, y se obtuvo el siguiente espectro, con una tolerancia de masa de 50 ppm:
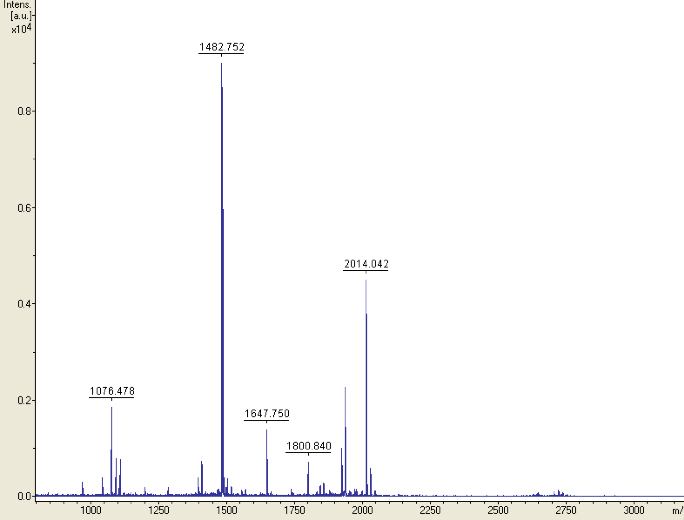
La lista de picos de masa que fueron etiquetados es la siguiente:
- 797.354
- 1041.570
- 1065.520
- 1076.478
- 1108.462
- 1235.560
- 1307.710
- 1365.640
- 1395.663
- 1482.752
- 1486.715
- 1647.750
- 1707.770
- 1800.840
- 2014.042
Pasos a seguir
-
Ingrese la lista de datos en el Mascot, y modifique los parámetros de búsqueda de acuerdo a los datos experimentales que se le dieron. Utilice como base de datos SwissProt, y por ahora mantenga la taxonomía en All entries. Evalúe la pantalla que sale como resultado (Concise Protein Summary).
-
El área en verde corresponde a asignaciones que no son consideradas significativas. ¿Hay algún hit que supera el threshold o umbral de identidad? ¿Cuál es? (anote el nombre y el número de acceso). Anote el Score que obtuvo. Anote también la versión de la base de datos utilizada y el tamaño de la base de datos utilizada en número de residuos.
-
¿Obtuvo algún hit por encima del umbral de identidad al utilizar la base de datos decoy?
-
Vaya al selector « Format as » y elija la pantalla Protein Summary. Fíjese los datos adicionales que aparecen en dicha pantalla. Vaya a Results List y analice la primera entrada. ¿Qué representa cada una de las columnas que se despliegan?
-
¿Hay algún péptido asignado con clivaje incompleto? Si los hay, identifique en cada caso cuál es el residuo que debería haberse cortado y no lo hizo.
-
¿Cuántos péptidos no asignados quedan para el primer hit? ¿A qué le parece que pueden corresponder?
-
Relance una búsqueda con los péptidos no asignados apretando el selector Search Unmatched. ¿Qué obtiene como resultado? ¿Qué puede deducir del mismo?
-
Si usted se fija en la lista de péptidos ingresados, puede identificar péptidos correspondientes a contaminación por queratinas. Hay programas de análisis que pueden reconocer esos péptidos contaminantes y eliminarlos de la tabla (p.ej. PeakErazor). Si hacemos esto, nos queda una lista de picos más reducida que es la siguiente:
- 1041.570
- 1076.478
- 1108.462
- 1395.663
- 1482.752
- 1486.715
- 1647.750
- 1800.840
- 2014.042
-
Relance la búsqueda con esta lista. ¿Cambió el primer hit? ¿Qué observa respecto al Score del primer hit? ¿Considera que el resto de los hits corresponden a proteínas mal identificadas?
-
Relance la búsqueda cambiando la base de datos a la del NCBInr. Anote la versión de la base de datos utilizada y el tamaño de la base de datos en número de residuos, así como el Score obtenido para el primer hit. Analice el Concise Protein Summary. ¿Obtiene los mismos resultados? ¿Dichos resultados son significativos? Con este resultado debería entender un poco más por qué la identificación de huella peptídica es probabilística…
-
Relance la búsqueda volviendo a elegir la base de datos SwissProt, pero evite agregarle la información sobre la carbamidometilación. ¿Qué sucede con la identificación? ¿Por qué le parece que sucede esto?
-
Dado que el material analizado proviene de líneas celulares humanas, relance la búsqueda ahora agregando la carbamidometilacion pero reduciendo la base de datos a la taxonomía Homo sapiens. ¿Qué observa en el Score de Mascot? ¿Qué observa en el Score del umbral o threshold? ¿Qué pasa con el Score para la base decoy? Anote los valores. ¿Cuál es la ventaja de restringir las especies o el grupo taxonómico en lugar de realizar una búsqueda de todos los grupos taxonómicos posibles?
-
En esta identificación, vaya a la lista del hits y pique el hipertexto LEG1HUMAN. Se abrirá una ventana independiente que se llama Protein View. Observe los datos que aparecen. Anote los datos adicionales que aparecen sobre la asignación de la proteína. ¿Qué opina del Sequence Coverage, considera que es bueno? Ahora que ve la secuencia completa de la proteína identificada, compruebe si la proteína tiene Cys que pudieran estar carbamiladas.
-
Pique el selector Show predicted peptides also. ¿Qué se muestra?. Usted, con estos datos, podría volver al espectro y analizar si es posible que haya más péptidos de esa proteína en el mismo, pero que no hayan sido etiquetados por alguna razon (p.ej. intensidad muy débil). Esta opción es muy útil para aumentar la confiabilidad en su medida.
-
Analice el gráfico de errores mostrado. En general se asume que es bueno que los errores se distribuyan alrededor del 0. ¿Es así en este caso? ¿Qué opina de la precisión y la exactitud de las medidas experimentales realizadas?
-
Fíjese en el gráfico o en la tabla cuál es el error máximo cometido en esta asignación. ¿Le parece que en función de esa observación se podría corregir el dato de peptide tolerance ingresado en la búsqueda original?
-
Relance la búsqueda considerando una peptide tolerance de 25 ppm. ¿Cómo se modifica el Score y el umbral de identidad? Haga lo mismo para una tolerancia de 1 Da. ¿Qué le dicen estas observaciones respecto a la importancia de la asignación correcta de la tolerancia de masa?
-
Resumiendo lo visto hasta el momento, ¿qué conclusión puede llegar respecto a la identidad de la proteína en cuestión? Entre otras cosas, observe los picos no asignados. ¿Que podrá pasar con esos picos?
-
Usted decide probar si alguno de los péptidos no asignados puede corresponder a péptidos de la proteína identificada que lleven modificaciones postraduccionales no previstas. La lista de péptidos no asignados es la siguiente:
- 1108.462
- 1395.663
- 1800.840
- 2014.042
-
Para ello, vaya al sitio del programa Find Mod http://www.expasy.ch/tools/findmod/
-
Alli inserte el ID de la proteína identificada (pruebe con LEG1_HUMAN), o copie e inserte la siguiente secuencia: MACGLVASNLNLKPGECLRVRGEVAPDAKSFVLNLGKDSNNLCLHFNPRFNAHGDANTIVCNSKDGGAWGTEQREAVFPFQPGSVAEVCITFDQANLTVKLPDGYEFKFPNRLNLEAINYMAADGDFKIKCVAFD
-
Inserte los picos no asignados en el lugar correspondiente. Elija todas las demas opciones como default, excepto las siguientes: WITH CYSTEINS TREATED WITH Iodoacetamide MASS TOLERANCE 50 ppm ALLOW FOR 1 MISSED CLEAVAGE SITES Presione Start Find Mod; cuando aparecen dos opciones de secuencia elija la Precursor y espere el resultado. ¿Se asignó algún péptido adicional? ¿Qué opina de la confiabilidad de la asignación?
-
Efectivamente, una de las modificaciones variables más comunes es la oxidación de metioninas, que suele darse por exposición al oxígeno del aire, especialmente cuando los péptidos digeridos se siembran en la placa de MALDI. Vuelva a largar la búsqueda del Mascot agregando la opción Oxidation (M) en Variable Modifications. ¿Qué resultados tiene? ¿Mejoró la identificación? Anote el nuevo Score.
IDENTIFICACION DE UNA PROTEÍNA DE UN SPOT DE GEL 2D POR COMBINACIÓN DE HUELLA PEPTÍDICA (PMF) Y ESPECTRO DE FRAGMENTACIÓN (PFF)
Gracias a las capacidades MS/MS del MALDI-TOF/TOF, usted puede contar con el espectro de fragmentación del pico de m/z 1647, el cual fue asignado en la huella peptídica. El mismo se ve de la siguiente manera:
Pico 1647
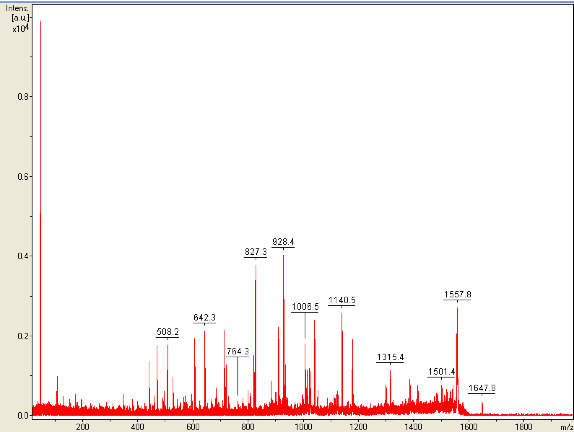
| FRAGMENTOS DEL PICO 1647 (lista parcial) |
|---|
| 442.2 |
| 470.2 |
| 491.3 |
| 495.2 |
| 508.2 |
| 527.2 |
| 544.3 |
| 570.3 |
| 572.2 |
| 596.3 |
Vamos a procesar esta información usando Mascot:
En el sitio de Matrix Science, pique el hipertexto Mascot. Le aparecerá una pantalla con opciones. Elija la que dice Mascot MS/MS Ion Search. Aquí usted podrá ingresar los datos de fragmentación de un determinado péptido proveniente de la huella peptídica, denominado ion precursor o parental. Se debe largar una búsqueda diferente para cada ion fragmentado, en este caso una para el pico 1647.750. La pantalla para procesar datos de fragmentación es similar a la de datos de huella peptídica. Sin embargo, hay algunos datos que se modifican, a saber:
- Las listas de masas moleculares de fragmentos deben subirse con un formato especial, no pastearse. En nuestro caso, las medidas se hicieron con un MALDI-TOF/TOF de Bruker, por lo cual ustedes cuentan con un archivo XML para ser usado en esta parte del ejercicio. El mismo debe ser cargado con el selector Data file, y especificado su formato con el selector Data format.
- Se debe indicar la m/z experimental del precursor en el campo Precursor, y la carga con la que viene el ion precursor en el campo Peptide charge. En este caso, como es un dato proveniente de MALDI, la carga es +1.
- Quantitation: si hay datos de cuantificación deben aclararse allí. Aquí no corresponde usar esta opción.
- Peptide tolerance es la tolerancia que corresponde a la medida del péptido parental o precursor, de la huella peptídica. Por ende, sigue siendo igual que en los ejercicios anteriores: 50 ppm. MS/MS tolerance es la correspondiente a la medición de los fragmentos derivados de dicho péptido. En general esta siempre es más alta, en este caso 1.2 Da. #C13 será de 0 si la asignacion de masa del peptido se hizo sobre el pico monoisotópico. En este caso es así, en algunas configuraciones de MS/MS se puede fragmentar otro peptido.
- Instrument: dado que cada tipo de instrumento tiene una forma de fragmentación propia de su configuración, el programa buscará las series adecuadas para cada configuración. Por ello se debe especificar el instrumento utilizado (en este caso MALDI-TOF/TOF).
- Error tolerant: esta es una opción que sólo está disponible en un análisis MS/MS. Si la cajita está tildada, se combinarán los resultados de dos búsquedas sucesivas: una búsqueda realizada con los parámetros ingresados por el usuario, y sobre los resultados de ella, una segunda búsqueda en la que el programa permite algunas excepciones a los parámetros iniciales (mayor tolerancia al error).
Pasos a seguir
- Vamos a largar una primera búsqueda para el ion precursor 1647. Esta búsqueda la haremos tildando Error tolerance. Tarda, ¿no? ¿Qué es lo que comprueba al ver el resultado? ¿Es la misma proteína identificada por huella peptídica? Anote el Score y el umbral de identidad. ¿Le parecen valores confiables?
- La pantalla que está mirando es la correspondiente al Peptide Summary. Fíjese qué se observa en la Protein View (picando el nombre de la proteína identificada) y en la Peptide View (picando el número que precede al péptido asignado). En esta última, fíjese cómo realizó el programa la asignación de las series de iones. ¿Están completas las asignaciones de la serie b y la y?
- Vaya al Format as y elija el formato Select Summary (unassigned). ¿Hay algún pico experimental no asignado?
- ¿Qué conclusión puede sacar respecto a la confiabilidad de la identificación por espectrometría de masas? ¿Qué Score es el que finalmente informaría a la comunidad científica como resultado de su identificación?
INFORME HUELLA PEPTíDICA
Redacte un texto que informe los resultados obtenidos en el experimento. Puede utilizar las imágenes y tablas de los resultados de las búsquedas para ilustrar los resultados. Informe los scores (y significancia) que considere relevantes. Debe incluir un segmento de metodología y también citar apropiadamente las herramientas y base de datos utilizadas. Máximo: 600 palabras
Preguntas orientadoras: como se obtuvo el spot? Como se trataron las proteínas? Parámetros importantes utilizados en la búsqueda en MASCOT? Cuantos péptidos asignados o no asignados? Porque? Hubo contaminaciones? Cobertura de la proteína? Cuales son las evidencias que tiene de que la proteína es la que Ud. dice? Cuanta confianza tiene en los resultados obtenidos?
PARA DISCUTIR Y ENTENDER
Que beneficios genera quitar los picos contaminantes?
Cual es la importancia de poner las modificaciones (fijas o variables) correctas? Que pasa si pongo de menos? Que pasa si pongo de mas?
Cual es la importancia de la tolerancia del error? En que cambia?
Que significa que estamos haciendo una identificación probabilística?
ANEXO 1 – Listas de las modificaciones postraduccionales más frecuentes y sus efectos sobre la determinación de masas moleculares