Trabajo Práctico 2 - Introducción a la anotación de variantes genómicas

Introducción
En este trabajo práctico haremos un repaso del pipeline bioinformático para el análisis de datos de NGS, incluyendo la anotación e interpretación de las variantes genómicas encontradas.
Dado que los pasos para el análisis de los datos ha sido visto en la materia Bioinformática/Análisis computacional de secuencias, en este trabajo nos centraremos en mayor medida en el paso de anotación de las variantes y en su interpretación en el contexto clínico. Existen diversos pipelines y programas para llevar a cabo este proceso, para este trabajo realizaremos la anotación funcional de variantes con el software libre snpEff, uno de los más utilizados en el mundo (y desarrollado por un argentino, Pablo Cingolani).
¿En qué consiste la anotación de variantes?
Una vez que obtenemos las variantes genómicas de la muestra de interés con respecto al genoma de referencia (llamado de variantes o variant calling), el siguiente paso es agregar información funcional a estás variantes para poder interpretar su rol en el contexto clínico que estamos estudiando. La inmensa mayoría de las variantes no van a estar directamente relacionadas con el fenotipo que estams estudiando, por lo tanto este paso es crucial para poder priorizar las variantes candidatas. Además, en este paso agregaremos información genómica de diversas bases de datos en las cuales podría estar reportada cada variante (gnomAD, 1000 genomas, ClinVar, dbSNP, etc.).
Para esto se utilizan diversos programas bioinformáticos que realizan una búsqueda para cada variante en distintas bases de datos conocidas y agregan la información encontrada. Además, estos programas suelen incorporar herramientas que permiten el filtrado y priorización de las variantes en base a la información de sus anotaciones.
Objetivos
- Como primper paso, repasaremos el pipeline para el análisis de datos de NGS, desde los FASTQ hasta el llamado de variantes. Con motivo de disminuir los tiempos de procesamiento y el poder computacional requerido, realizaremos el análisis de una muestra de ADN mitocondrial humano (mtDNA).
- Luego, utilizaremos archivos VCF pre-generados de una familia (CEPH1463) provenientes de la base de datos de 1000 genomas, en donde se hizo el llamado de variantes de los 3 individuos con respecto al genoma de referencia humano GRCh38. Utilizaremos este archivo como punto de partida para la anotación y filtrado de variantes.
- Por último, cada grupo analizará un archivo VCF correspondiente a padre, madre e hija de la misma familia CEPH1463 y deberá encontrar la variante responsable de la enfermedad indicada en cada caso . Si bien todos los grupos estudiarán a los mismos 3 individuos, los datos fueron modificados manualmente para incorporar distintas variantes asociadas a enfermedades que cada grupo deberá encontrar.
Ejercicio 1: Alineamiento
Para mapear nuestras lecturas de mtDNA al genoma mitocondrial de referencia, vamos a recurrir a BWA-MEM2, la última versión de un alineador ampliamente utilizado que tiene la ventaja de ser rápido y a su vez eficiente en relación a la cantidad de memoria que utiliza.
Nota: Para utilizar los siguientes programas debemos activar el environment genomica_uade de conda en la máquina virtual.
- Primero realizamos el control de calidad de los archivos fastq con fastqc
Creamos una carpeta para generar el reporte:
mkdir FASTQC
fastqc -o ./FASTQC/ -t 2 ./reads/sra_data.fastq
-
¿Cómo es la calidad de los datos? ¿Haría algúna corrección?
-
Antes de alinear las lecturas hay que crear un índice del genóma de referencia, también llamado “indexar”. Para eso, parados en la carpeta
TP2, ejecutamosbwa-mem2 index:
Búsqueda: ¿Qué significa indexar un genoma de referencia?
bwa-mem2 index ./reference_genome/Homo_sapiens.GRCh37.74.dna.chromosome.MT.fa
- Luego, siempre parados en la carpeta
TP2, alineamos las lecturas al genoma de referencia, resultando en un archivo SAM que redirigimos a samtools para que lo ordene por coordenadas y lo convierta en bam.
bwa-mem2 mem -t 2 ./reference_genome/Homo_sapiens.GRCh37.74.dna.chromosome.MT.fa ./reads/sra_data.fastq | samtools sort -@2 -m2g -o ./sra_data_sorted.bam
Búsqueda: ¿Qué implica el sorting de un archivo bam? ¿Qué quiere decir que un archivo bam/sam esté ordenado por coordenadas?
- Inspeccione el archivo bam:
samtools view sra_data_sorted.bam | head
-
¿Por qué no se puede ver directamente con head?
-
Ahora podemos usar qualimap para evaluar el alineamiento.
qualimap bamqc -bam ./sra_data_sorted.bam -gd HUMAN -nt 2
- En base al reporte, ¿Cómo puede decir que fue al alineamiento? ¿Cuántas lecturas alinearon a la referencia?
Ejercicio 2: Visualización de datos de secuenciación en IGV
Para visualizar el mapeo de las lecturas contra el genoma de referencia vamos a utilizar el programa IGV (Integrative Genomics Viewer).
- Primero, necesitamos indexar el archivo bam ordenado anteriormente:
samtools index sra_data_sorted.bam
-
Parados en la carpeta
TP2como siempre, chequeamos si están generados los archivos que necesitamos conls -l. -
Abrimos IGV
IGV se abre escribiendo
igv+ enter en la terminal.
Primero, debemos importar el genoma de referencia y su anotación, para visualizar los genes y sus localizaciones:
-
En el menú superior de IGV, vamos a Genomes -> Create Genome File, completamos los campos con el nombre que deseemos.
-
En el campo FASTA file cargamos el archivo que contiene el genoma mitocondrial de referencia
Homo_sapiens.GRCh37.74.dna.chromosome.MT.faque se encuentra en la carpetaTP2/reference_genome -
En el campo Gene file cargamos el archivo de anotación
HS.MT.gtfque también se encuentra en la carpeta mencionada. Creamos entonces el Genome file en IGV. -
A continuación, desde el menú File -> Load from file cargamos el archivo con nuestras lecturas en formato bam ordenado, llamado
sra_data_sorted.bam.
Es importante que el archivo index o índice que creamos en el paso 1 (.bai) se encuentre en la misma carpeta que el sorted .bam
- ¿Qué información nos muestra a priori IGV?
- Coloreamos las lecturas de acuerdo a si corresponden a la hebra forward o reverse: click derecho en cualquier lectura y Color alignments by -> read strand
- Ordenamos las lecturas por forward o reverse: click derecho en cualquier lectura y Sort alignments by -> read strand, click derecho en cualquier lectura y Order alignments by -> read strand
- Ahora vamos a describir parámetros que nos pueden ayudar a ver la calidad de la secuenciación y detectar tanto posibles variantes genéticas verdaderas, como posibles falsos positivos. Registren los parámetros mencionados y en qué sección de IGV se informan.
Ejercicio 2.1: Llamado de variantes (variant calling)
Ahora que ya tenemos las lecturas alineadas contra la referencia podemos proceder con el llamado de variantes o variant calling. Hay diversos algoritmos y programas para hacerlo, en este caso vamos a usar FreeBayes, que utiliza un método bayesiano y basado en haplotipos para poder detectar variantes.
Si bien el programa admite distintas opciones para correrlo, usaremos el comando básico con todos los parámetros por default. Como siempre, pueden ver la ayuda del programa con freebayes –help o accediendo a la documentación del mismo online ( https://github.com/freebayes/freebayes).
Para ejecutarlo con su configuración básica solo debemos pasarle el archivo fasta de la referencia y el archivo bam que generamos durante el alineamiento. El programa comparará las lecturas alineadas con la secuencia de la referencia para detectar variantes y asignarle scores de calidad según el contexto en el que se encuentren.
freebayes -f ./reference_genome/Homo_sapiens.GRCh37.74.dna.chromosome.MT.fa sra_data_sorted.bam > MT_alineamiento.vcf
El resultado del variant calling será el archivo vcf (MT_alineamiento.vcf) conteniendo las variantes detectadas.
- Observe el archivo VCF y comente algunas de las variantes encontradas.
- ¿Cuántas variantes detectó FreeBayes?
Ejercicio 3: Anotación y filtrado de variantes
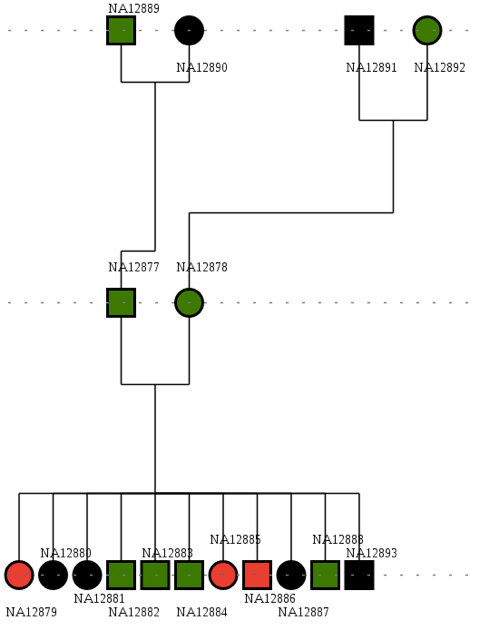
Se analizará una familia de 17 individuos como se ve en la imagen del pedigree. De toda la familia, hay 3 individuos de la última generación que tienen fibrosis quística y se debe encontrar la variante responsable de la enfermedad recesiva (estos individuos no estaban realmente enfermos, se agregó manualmente la variante causante de la enfermedad para generar este dataset de prueba). Para acelerar los tiempos y para que se pueda hacer el análisis en computadoras personales se utilizará solo un VCF con las variantes del cromosoma 7 y el 17.
Nota: El alineamiento y el llamado de variantes se realizó con el genoma de referencia humano GRCh37
- Primero debemos copiar la carpeta snpEff que descargamos en nuestra carpeta home:
cp -r Archivos_para_TP1_bis/snpEff ~/
- Luego, nos moveremos a la carpeta Intro para comenzar a trabajar:
cd Archivos_para_TP1_bis/Intro
- Anotación primaria de variantes (predicción de pérdida de función, tipo de variante, cambio de aminoácidos, gen afectado)
java -Xmx4g -jar ~/snpEff/snpEff.jar -v -stats ex1.html GRCh37.75 ./ex1.vcf > ./ex1.ann.vcf
Esto genera un archivo html con el reporte del análisis y un nuevo VCF con las variantes anotadas. Observar el reporte y el nuevo VCF.
-
Contar las variantes en casos y controles (no correr este comando porque lleva mucho tiempo, tienen el archivo ya generado en la carpeta)
En este paso, el comando caseControl va a contabilizar, para cada variante en el VCF, cuántos de los individuos casos (enfermos) son homocigotas y heterocigotas para esa variante, y cuántos controles (no enfermos). El formato en el que se anota esta información es por ejemplo:
Cases=1,1,3oControls=8,6,22, siendo el primer valor la cantidad de homocigotas, el segundo la de heterocigotas y el tercero el conteo total de alelos con esa variante (recuerden que el homocigota tendrá 2 alelos). Esto nos servirá para después poder filtrar las variantes de acuerdo a lo que estamos buscando. Para que el programa sepa quiénes son casos y controles se le pasa un archivo .tfam que tiene esta información (para ver el formato de este archivo: https://www.cog-genomics.org/plink/1.9/formats#tfam)
java -Xmx1g -jar ~/snpEff/SnpSift.jar \
caseControl \
-v \
-tfam varios/pedigree.tfam \
./ex1.ann.vcf \
> ./ex1.ann.cc.vcf
Este paso tarda bastante en correr así que ya tienen el archivo resultante en la carpeta Archivos_para_TP1_bis/varios/
- Filtrado de variantes: Como estamos buscando una variante recesiva, queremos quedarnos con variantes que estén en homocigosis en los casos pero no en los controles. Además buscaremos variantes que tengan un impacto predicho de Alto o Moderado. Para eso filtraremos el VCF de la siguiente manera:
cat ./varios/ex1.ann.cc.vcf \
| java -jar ~/snpEff/SnpSift.jar filter \
"(Cases[0] = 3) & (Controls[0] = 0) & ((ANN[*].IMPACT = 'HIGH') | (ANN[*].IMPACT = 'MODERATE'))" \
> ./ex1.filtered.vcf
- Para visualizar las variantes resultantes:
cat ./ex1.filtered.vcf | ~/snpEff/scripts/vcfInfoOnePerLine.pl
¿Qué observan?
- Anotación con ClinVar: En este caso la variante causante de la enfermedad es en realidad ya conocida. Si le agregamos al VCF la anotación de ClinVar podremos ver la información asociada a fibrosis quística en una de las variantes a la que habíamos llegado en el paso anterior:
java -Xmx1g -jar ~/snpEff/SnpSift.jar \
annotate \
-v \
./varios/db/clinvar.vcf.gz \
./varios/ex1.ann.cc.vcf \
> ./ex1.ann.cc.clinvar.vcf
Nota: Tener en cuenta que como este VCF fue generado usando como genoma de referencia el GRCh37 la base de datos de ClinVar a utilizar debe corresponder a la misma versión, ya que si no las coordenadas genómicas de las variantes pueden variar. En este caso el archivo clinvar.vcf.gz contiene la versión GRCh37.
- Ver variantes con anotación de ClinVar = “Cystic Fibrosis”:
cat ./ex1.ann.cc.clinvar.vcf \
| java -jar ~/snpEff/SnpSift.jar filter \
"(exists CLNDBN) & (ANN[*].EFFECT has 'stop_gained') & (ANN[*].GENE = 'CFTR')" \
> ./ex1.ann.cc.clinvar.filtered.vcf
Ejercicio 4: Búsqueda de variante asociada a una enfermedad
Cada grupo deberá utilizar el VCF y el pedigree dentro de la carpeta correspondiente para anotar y filtrar las variantes que le permitan llegar a la/las candidatas según el modelo de herencia de la enfermedad:
Nota: El alineamiento y el llamado de variantes en este caso se realizó con el genoma de referencia humano GRCh38. Por lo tanto, deberá utilizar las bases de datos correspondientes para hacer la anotación. Ayuda: la base de datos que usa SnpEff para esta versión del genoma de referencia es la GRCh38.99.
- Grupo 1 - Fenilcetonuria
- Autosómica Recesiva, enferma la hija y no los padres
- Grupo 2 - Anemia falciforme
- Autosómica Recesiva, enferma la hija y no los padres
- Grupo 3 - Albinismo oculocutáneo
- Autosómica Recesiva, enferma la hija y no los padres
- Grupo 4 - Hipercolesterolemia
- Autosómica Dominante, enfermos los padres y no la hija.
- Grupo 5 - Neurofibromatosis tipo 1
- Autosómica Dominante, enfermas madre e hija y no el padre.
- Grupo 6 - Enfermedad renal poliquística.
- Autosómica Dominante, enfermos padre e hija y no la madre.
- Grupo 7 - Síndrome de Rett
- Dominante ligada al X, padres no enfermos y la hija sí.
Bibliografía
- Muestra descargada del Sequence Read Archive ( http://trace.ncbi.nlm.nih.gov/Traces/sra/?run=DRR001063)
- https://github.com/bwa-mem2/bwa-mem2
- Samtools ( http://www.htslib.org/)
- FreeBayes https://arxiv.org/abs/1207.3907
- IGV ( http://software.broadinstitute.org/software/igv/)
- Paper del estudio de genomas mitocondriales del cual obtuvimos la muestra para el TP ( https://genomebiology.biomedcentral.com/track/pdf/10.1186/gb-2011-12-6-r59)
- SnpEff y tutorial: https://pcingola.github.io/SnpEff/
- Muestras del proyecto 1000 genomas: http://hgdownload.soe.ucsc.edu/gbdb/hg38/1000Genomes/trio/NA12878_1463_CEU/ y https://www.completegenomics.com/public-data/