Trabajo Práctico 1 - Introducción al ensamblado de secuencias genómicas y visualización en IGV

Introducción
-
El ensamblado de secuencias de ADN se refiere al proceso de alineamiento y fusión de fragmentos que se originaron a partir de una secuencia de ADN más larga, para reconstruir esa secuencia original. Esto es necesario ya que la mayoría de las tecnologías de secuenciación de ADN no pueden leer genomas completos de una sola vez, sino que leen pequeños fragmentos menores de tamaño variable, dependiendo de la tecnología utilizada. Por lo general, los fragmentos cortos, llamados lecturas (reads), suelen ser el resultado de haber fragmentado inicialmente el ADN que se quería secuenciar, o de seleccionar regiones del mismo por métodos tales como PCR o captura con sondas.
-
El proceso de ensamblar una secuencia se puede comparar con tomar muchas copias de un libro, recortar estas copias en fragmentos diferentes e intentar recomponer el texto del libro con sólo mirar las piezas recortadas. Además de la dificultad obvia de esta tarea, hay algunos problemas prácticos adicionales: el original puede tener muchos párrafos repetidos, y algunos fragmentos pueden modificarse durante la destrucción para tener errores tipográficos. También se pueden agregar extractos de otro libro, y algunos fragmentos pueden ser completamente irreconocibles.
-
En el ensamblado de secuencia, se pueden distinguir dos tipos diferentes:
Ensamblado de novo: consiste en ensamblar lecturas cortas para crear secuencias completas (a veces novedosas), sin usar un genoma de referencia. Esta estrategia se utiliza cuando el organismo de interés todavía no podee un genoma consenso o de referencia, o si lo posee éste no es de buena calidad.
Mapeo: involucra ensamblar lecturas tomando una secuencia principal existente, denominada genoma de referencia. Tiene como resultado la construcción de una secuencia que es muy similar pero no necesariamente idéntica a la secuencia de referencia.
- En términos de complejidad y requisitos de tiempo, los ensamblados de novo son varios órdenes de magnitud más lentos y requieren más memoria que los mapeos contra genoma de referencia. Esto se debe principalmente al hecho de que el algoritmo de ensamblado de novo necesita comparar cada lectura con cualquier otra lectura del conjunto analizado.
Objetivos
- A partir de una muestra de ADN mitocondrial humano (mtDNA), generar un ensamblado de novo y un mapeo contra genoma mitocondrial de referencia.
- Visualizar archivos de lecturas alineadas y comprender los conceptos de: profundidad de lectura, cobertura horizontal, calidad de mapeo, calidad de base, falsos positivos, entre otras cuestiones que permite la visualización de estos archivos.
Ejercicio 1: Ensamblado de novo y evaluación
Hoy vamos a ensamblar de novo un genoma mitocondrial humano utilizando ABySS, un ensamblador de ejecución sencilla.
Para eso utilizaremos una corrida de Illumina single-reads obtenida del repositorio de NCBI llamado Sequence Read Archive.
Todo lo necesario para realizar este TP se encuentra en la máquina virtual entregada, y todos los archivos de este TP se encuentran en la carpeta
/home/alumno/TPs/TP1.
El archivo
.fastqcon las lecturas está en/home/alumno/TPs/TP1/reads/sra_data.fastq.
- En este TP usaremos línea de comando. Para eso abrimos la terminal (que ya nos ubica dentro de
alumno) y vamos a la carpeta TP1. Inspeccionemos también el encabezado de nuestro archivo .fastq. ¿Qué contiene?
cd TPs/TP1/
head ./reads/sra_data.fastq
- Veamos cómo es el script que utilizaremos para Abyss, que es un archivo de texto con extensión .sh. También tendremos que eliminar un formato de fin de línea propio de Windows para que la terminal en Ubuntu lo pueda leer correctamente:
cat abyss_script.sh
sed -i -e 's/\r$//' abyss_script.sh
- ¿Qué hace el comando seq? ¿Y el bucle for?
Recordemos lo aprendido en la materia Bioinformática (o refresquemos con Google).
- Usando el comando
mkdir, creamos una carpeta llamadaabyssen la carpetaTP1:
mkdir abyss
- Ejecutamos el script para ABySS:
./abyss_script.sh
- ¿Qué sucede si ejecutamos
ls -l ./abyss/contigs-k*.fa?
El asterisco (
*) significa que en esa posición del nombre del archivo .fa puede haber 0 o más caracteres. Para más información pueden buscar en Google wildcard operators, o regular expressions
- Con ABySS se puede hacer una inspección rápida de los resultados obtenidos en los distintos ensamblados utilizando el comando
abyss-fac:
abyss-fac ./abyss/contigs-k*.fa
- Si se tiene una estimación del tamaño del genoma, se puede añadir también con el parametro
-epara obtener estadísticas asociadas.
El genoma mitocondrial humano tiene un tamaño aproximado de 16Kb (16569b para ser exactos):
abyss-fac -e 16000 ./abyss/contigs-k*.fa
Del manual de ABySS podemos obtener el significado de las métricas observadas:
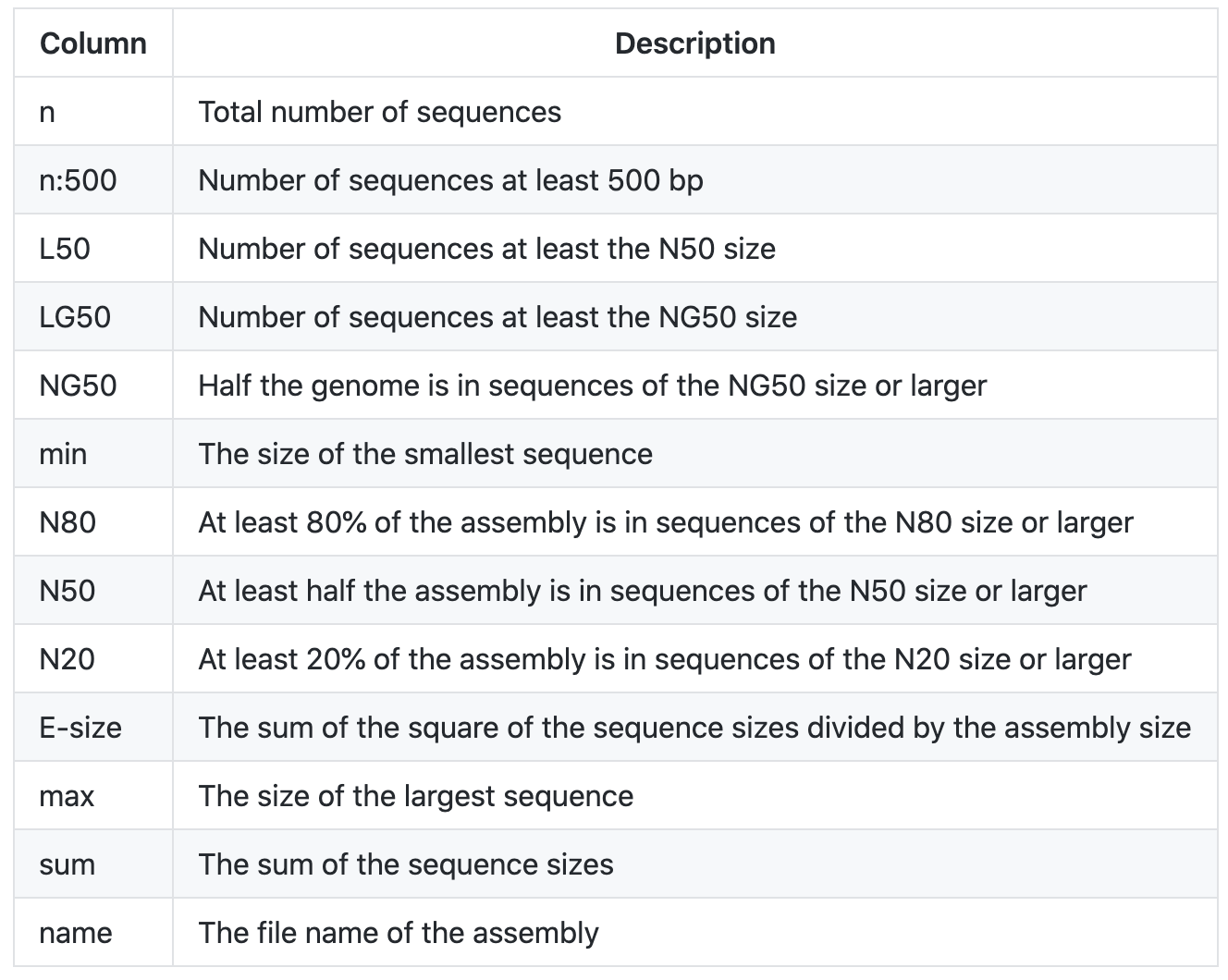
-
¿Cómo guardaría esos resultados en un archivo llamado
abyss_fac.rslts? -
En base a los resultados, ¿cuál podría ser el mejor ensamblado? ¿Por qué?
Ejercicio 2: Alineamiento a un genoma de referencia y visualización
Para mapear nuestras lecturas de mtDNA al genoma mitocondrial de referencia, vamos a recurrir a Bowtie2, un alineador ampliamente utilizado que tiene la ventaja de ser ultra rápido y a su vez eficiente en relación a la cantidad de memoria que utiliza.
- Primero hay que crear un índice del genóma de referencia, también llamado “indexar”. Para eso, parad@s en la carpeta
TP1, ejecutamosbowtie2-build:
Búsqueda: ¿Qué significa indexar un genoma de referencia?
Creamos carpeta bowtie dentro de TP1
mkdir bowtie
bowtie2-build ./reference_genome/Homo_sapiens.GRCh37.74.dna.chromosome.MT.fa ./bowtie/human_mito_ref
- Luego, siempre parad@s en la carpeta
TP1, alineamos las lecturas al genoma de referencia, resultando en un archivo SAM:
bowtie2 -x ./bowtie/human_mito_ref -U ./reads/sra_data.fastq -S alineamiento.sam
- ¿Cuántas lecturas alinearon a la referencia? Inspeccione el archivo .sam ejecutando el comando
head.
Ejercicio 3: Visualización de datos de secuenciación en IGV
Para visualizar el mapeo de las lecturas contra el genoma de referencia vamos a utilizar el programa IGV (Integrative Genomics Viewer).
- En primer lugar será necesario convertir nuestro alineamiento hecho en bowtie2 de formato .sam (Sequence Alignment/Map) a formato .bam (Binary Alignment Map), su versión binaria. Para eso utilizaremos el programa SAMtools, que proporciona herramientas para procesar esa clase de archivos:
samtools view -Sb alineamiento.sam > alineamiento.bam
- Luego el archivo debe ser ordenado y luego indexado:
Búsqueda: ¿Qué implica el sorting de un archivo bam?
samtools sort alineamiento.bam -o alineamiento.sorted.bam
samtools index alineamiento.sorted.bam
-
Parad@s en la carpeta
TP1como siempre, chequeamos si están generados los archivos que necesitamos conls -l. ¿Inspeccionamos el archivo bam resultante conhead? -
Abrimos IGV
IGV se abre escribiendo
igv+ enter en la terminal.
Primero, debemos importar el genoma de referencia y su anotación, para visualizar los genes y sus localizaciones:
-
En el menú superior de IGV, vamos a Genomes -> Create Genome File, completamos los campos con el nombre que deseemos.
-
En el campo FASTA file cargamos el archivo que contiene el genoma mitocondrial de referencia
Homo_sapiens.GRCh37.74.dna.chromosome.MT.faque se encuentra en la carpetaTP1/reference_genome -
En el campo Gene file cargamos el archivo de anotación
HS.MT.gtfque también se encuentra en la carpeta mencionada. Creamos entonces el Genome file en IGV. -
A continuación, desde el menú File -> Load from file cargamos el archivo con nuestras lecturas en formato bam ordenado, llamado
alineamiento.sorted.bam.
Es importante que el archivo index o índice que creamos en el paso 2 (.bai) se encuentre en la misma carpeta que el sorted .bam
-
¿Qué información nos muestra a priori IGV?
-
Coloreamos las lecturas de acuerdo a si corresponden a la hebra forward o reverse: click derecho en cualquier lectura y Color alignments by -> read strand
-
Ordenamos las lecturas por forward o reverse: click derecho en cualquier lectura y Sort alignments by -> read strand, click derecho en cualquier lectura y Order alignments by -> read strand
-
Ahora vamos a describir parámetros que nos pueden ayudar a ver la calidad de la secuenciación y detectar tanto posibles variantes genéticas verdaderas, como posibles falsos positivos. Registren los parámetros mencionados y en qué sección de IGV se informan.
Bibliografía
- ABySS ( http://www.bcgsc.ca/platform/bioinfo/software/abyss)
- Muestra descargada del Sequence Read Archive ( http://trace.ncbi.nlm.nih.gov/Traces/sra/?run=DRR001063)
- Bowtie2 ( http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
- Samtools ( http://www.htslib.org/)
- IGV ( http://software.broadinstitute.org/software/igv/)
- Paper sobre grafos y ensamblado de genomas ( https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5531759/pdf/nihms329513.pdf)
- Paper del estudio de genomas mitocondriales del cual obtuvimos la muestra para el TP ( https://genomebiology.biomedcentral.com/track/pdf/10.1186/gb-2011-12-6-r59)